Example workflow using FUSE
Susanna Holmstrom
2026-02-05
example.RmdReading in data
We start by reading in the data, which consist of the tables K0 and K1. Both have CpG sites as rows and samples as columns, and
- K0 contains the unmethylated counts
- K1 contains the methylated counts
This is a dummy data set consisting of manipulated counts of the 100 000 first CpG sites in chromosome 20.
k0_file <- system.file("examples/k0.tsv.gz", package = "methFuse")
k1_file <- system.file("examples/k1.tsv.gz", package = "methFuse")
K0 <- read.table(k0_file, header = TRUE)
K1 <- read.table(k1_file, header = TRUE)
head(K0)## Sample1 Sample2 Sample3 Sample4 Sample5 Sample6 Sample7 Sample8
## chr20.60008 0 6 0 0 0 0 0 0
## chr20.60009 0 0 9 0 17 0 17 0
## chr20.60119 21 0 0 22 0 21 0 3
## chr20.60120 17 0 0 21 16 19 0 0
## chr20.60578 0 16 0 8 5 0 0 19
## chr20.60579 0 13 17 0 10 0 0 11
## Sample9 Sample10 Sample11 Sample12 Sample13 Sample14 Sample15
## chr20.60008 0 17 18 0 7 0 0
## chr20.60009 17 7 1 19 3 16 0
## chr20.60119 0 21 6 0 0 17 16
## chr20.60120 0 0 19 0 0 0 11
## chr20.60578 0 4 0 4 15 0 0
## chr20.60579 0 2 20 0 0 16 6
## Sample16 Sample17 Sample18 Sample19 Sample20
## chr20.60008 17 13 17 21 0
## chr20.60009 16 10 0 16 0
## chr20.60119 3 2 15 9 0
## chr20.60120 0 0 5 18 0
## chr20.60578 19 0 0 22 0
## chr20.60579 0 2 0 0 9Apply FUSE as a pipeline
The whole FUSE pipeline can be applied using the function ‘fuse.segment()’. It performs the following steps:
- clustering
- cutting clustering tree optimally
- summarizing segments
The function takes as input the count matrices K0 and K1 and the chromosome and position information for each CpG site, and outputs a summary table and a data frame with betas.
segment_result <- fuse.segment(
as.matrix(K0),
as.matrix(K1),
chr = sub("\\..*$", "", rownames(K0)),
pos = as.integer(sub("^.*\\.", "", rownames(K0))),
method = "BIC"
)
head(segment_result$summary)## Segment Chr Start End CpGs Length Beta Coherent
## 1 chr20.60008 chr20 60008 61140 17 1133 0.8378468 FALSE
## 2 chr20.61141 chr20 61141 61817 12 677 0.8450438 FALSE
## 3 chr20.61818 chr20 61818 63862 18 2045 0.8514536 FALSE
## 4 chr20.63863 chr20 63863 64424 13 562 0.8565943 FALSE
## 5 chr20.64609 chr20 64609 64728 6 120 0.6418476 FALSE
## 6 chr20.64778 chr20 64778 65365 6 588 0.1449167 FALSE
head(segment_result$betas_per_segment)## Sample1 Sample2 Sample3 Sample4 Sample5 Sample6
## chr20.60008 0.7331288 0.7795786 0.8133333 0.84156977 0.7853659 0.81250000
## chr20.61141 0.8740920 0.7928571 0.8174157 0.86084906 0.8803612 0.89154013
## chr20.61818 0.8943894 0.8278388 0.7691083 0.89508197 0.8259587 0.82819383
## chr20.63863 0.8560748 0.8793103 0.8287154 0.87633262 0.7705628 0.81779661
## chr20.64609 0.5810056 0.7909605 0.5865385 0.63636364 0.6150943 0.71794872
## chr20.64778 0.2077922 0.1000000 0.2321429 0.02764977 0.0000000 0.07692308
## Sample7 Sample8 Sample9 Sample10 Sample11 Sample12
## chr20.60008 0.8653251 0.89727127 0.9042357 0.81441718 0.7964169 0.8934911
## chr20.61141 0.9152120 0.94017094 0.7426326 0.85650224 0.7752294 0.7946429
## chr20.61818 0.8787313 0.77701544 0.8374291 0.83053691 0.8900145 0.8762376
## chr20.63863 0.8160622 0.77941176 0.8909513 0.74315789 0.8958333 0.9621749
## chr20.64609 0.6912442 0.69565217 0.7898551 0.51933702 0.6346154 0.6311111
## chr20.64778 0.2844037 0.05769231 0.1984733 0.08024691 0.1188811 0.1530612
## Sample13 Sample14 Sample15 Sample16 Sample17 Sample18
## chr20.60008 0.8477930 0.85780526 0.8138848 0.8699187 0.8298755 0.89660743
## chr20.61141 0.9192140 0.91549296 0.8333333 0.8463357 0.8592965 0.78795812
## chr20.61818 0.8681481 0.81139756 0.8013468 0.8629283 0.8820059 0.93140244
## chr20.63863 0.8687783 0.83193277 0.8916479 0.9232614 0.8540434 0.80167015
## chr20.64609 0.5294118 0.66666667 0.6567164 0.4928910 0.6908213 0.65024631
## chr20.64778 0.1682692 0.05462185 0.2631579 0.1298077 0.2117117 0.05952381
## Sample19 Sample20
## chr20.60008 0.7971014 0.9366603
## chr20.61141 0.7953092 0.8556034
## chr20.61818 0.9240924 0.8153619
## chr20.63863 0.9120172 0.9628713
## chr20.64609 0.6808511 0.6701031
## chr20.64778 0.2166667 0.2110092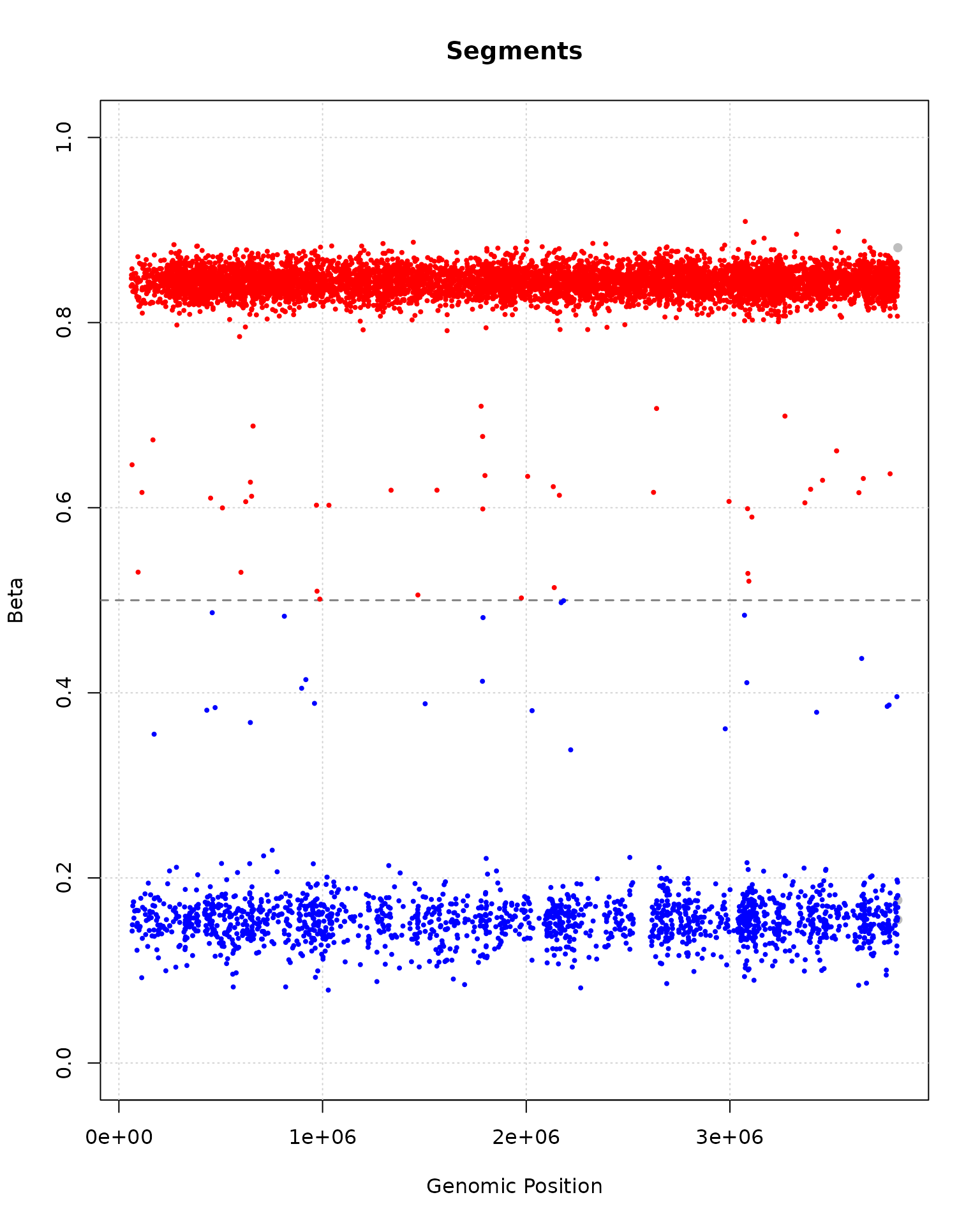
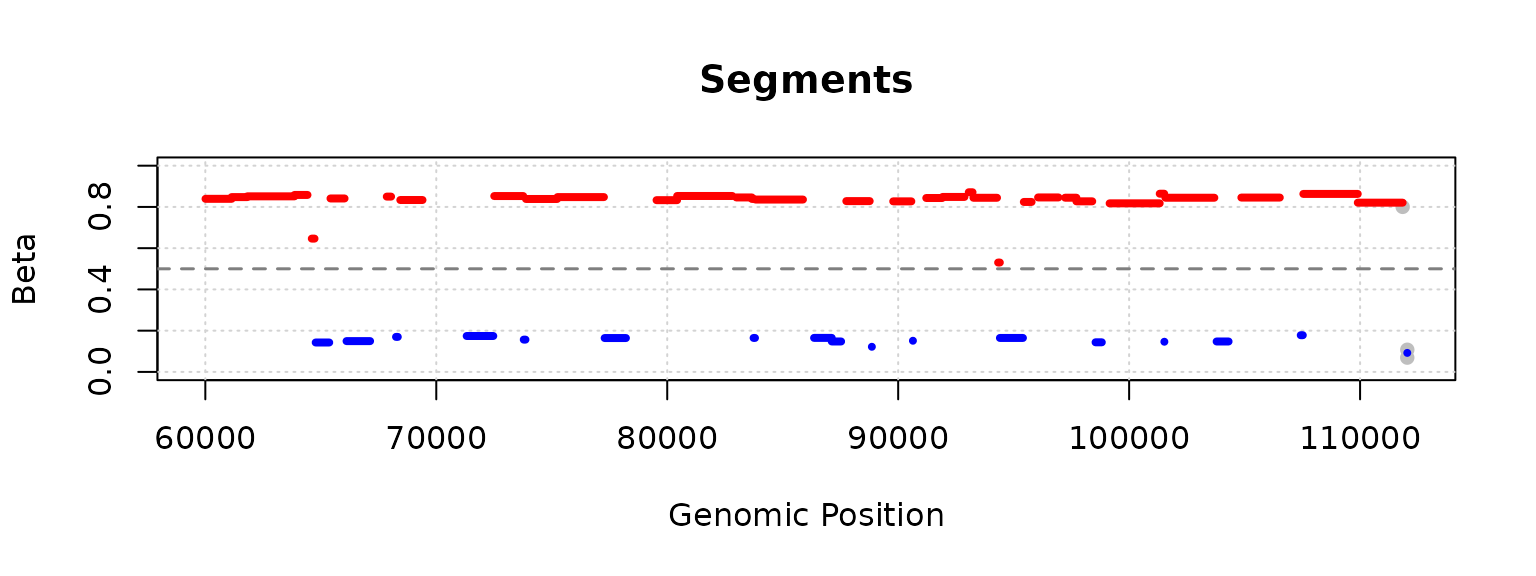
plot(segment_result, segments_to_plot = 50:60)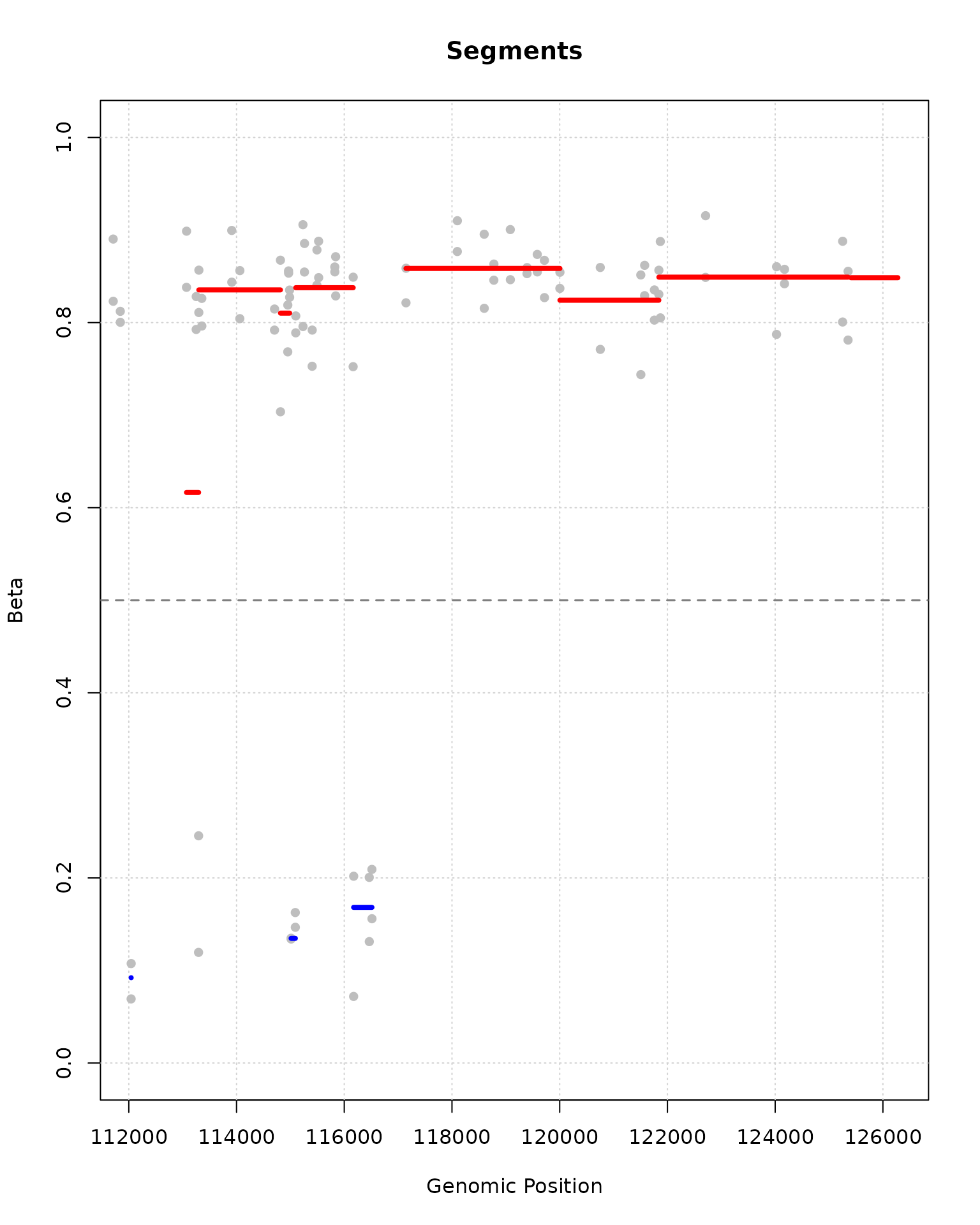
Using BSseq or methrix
The ‘fuse.segment()’ function accepts as input also a BSseq or a methrix object.
library(bsseq)
set.seed(1)
M <- matrix(c(sample(0:10, 1000, TRUE),
sample(50:60, 1000, TRUE),
sample(0:10, 1000, TRUE),
sample(20:30, 1000, TRUE))
, nrow = 1000, byrow = T)
Cov <- M + matrix(sample(50:60, 4000, TRUE), nrow = 1000)
bs <- BSseq(
chr = rep("chr1", 1000),
pos = seq_len(1000),
M = M,
Cov = Cov,
sampleNames = colnames(M)
)
res <- fuse.segment(bs)
head(res$summary)## Segment Chr Start End CpGs Length Beta Coherent
## 1 chr1.1 chr1 1 250 250 250 0.08344157 FALSE
## 2 chr1.251 chr1 251 500 250 250 0.50035855 TRUE
## 3 chr1.501 chr1 501 750 250 250 0.08405464 FALSE
## 4 chr1.751 chr1 751 1000 250 250 0.31483887 TRUE
library(methrix)
data(methrix_data, package = "methrix")
res <- fuse.segment(methrix_data)
head(res$summary)## Segment Chr Start End CpGs Length Beta Coherent
## 1 chr21.27866423 chr21 27866423 47286854 82 19420432 0.09796720 TRUE
## 2 chr21.47286962 chr21 47286962 47517454 118 230493 0.16972343 TRUE
## 3 chr21.47517456 chr21 47517456 47520741 211 3286 0.05453577 TRUE
## 4 chr21.47536415 chr21 47536415 47540455 129 4041 0.11761019 TRUE
## 5 chr22.45083239 chr22 45083239 49007398 203 3924160 0.13991860 TRUEApply FUSE through separate steps
If the intermediate outputs of the method are relevant, then the pipeline can also be applied by calling each of the functions ‘fuse.cluster()’, ‘number.of.clusters()’, ‘fuse.cut.tree()’, and ‘fuse.summary()’ separately.
1. Cluster
In the first step, ‘fuse.cluster()’ is applied on the count matrices. This performs a hierarchical clustering of closest neighbors, and outputs a clustering tree of class ‘hclust’.
tree <- fuse.cluster(as.matrix(K0),
as.matrix(K1),
chr = sub("\\..*$", "", rownames(K0)),
pos = as.integer(sub("^.*\\.", "", rownames(K0))))
names(tree)## [1] "merge" "height" "order" "labels" "call"
## [6] "method" "dist.method"
head(tree$merge)## [,1] [,2]
## [1,] -45579 -45580
## [2,] -57737 -57738
## [3,] -46190 -46191
## [4,] -49658 -49659
## [5,] -23937 -23938
## [6,] -41560 -41561
head(tree$height)## [1] 10.65993 23.28925 35.99633 48.57419 65.21045 82.47522The clustering tree contains the following elements:
- ‘merge’: the labels of the merged points
- ‘height’: The total log-likelihood for forming this merge
- ‘order’: Same order as original, since clustering is done in 1D
- ‘labels’: Labels of form ‘chr.pos’
- ‘call’: ‘fuse.cluster(K0, K1)’
- ‘method’: ‘fuse’
- ‘dist.method’: ‘fuse’
2. Cutting the tree
In order to cut the clustering tree, the optimal number of segments needs to be calculated. For this we have the function ‘number.of.clusters()’, which performs model selection by minimizing either the Bayesian Information Criterion (BIC, default), or the Akaike Information Criterion (AIC).
optimal_num_of_segments <- number.of.clusters(tree, ncol(K0), 'BIC')
optimal_num_of_segments## [1] 9030The tree can then be cut using ‘fuse.cut.tree()’, (or ‘cutree’ from package ‘stats’).
segments <- fuse.cut.tree(tree, optimal_num_of_segments)
head(segments, 100)## [1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 3 3 3 3 3 3 3 3
## [38] 3 3 3 3 3 3 3 3 3 3 4 4 4 4 4 4 4 4 4 4 4 4 4 5 5 5 5 5 5 6 6 6 6 6 6 7 7
## [75] 7 7 7 7 7 7 7 7 7 7 8 8 8 8 8 8 8 8 8 8 8 8 8 8 9 93. Summarizing segments
The segments can be summarized in a table with the function ‘fuse.summary()’.
result <- fuse.summary(as.matrix(K0),
as.matrix(K1),
chr = sub("\\..*$", "", rownames(K0)),
pos = as.numeric(sub("^.*\\.", "", rownames(K0))),
segments)
head(result$summary)## Segment Chr Start End CpGs Length Beta Coherent
## 1 chr20.60008 chr20 60008 61140 17 1133 0.8378468 FALSE
## 2 chr20.61141 chr20 61141 61817 12 677 0.8450438 FALSE
## 3 chr20.61818 chr20 61818 63862 18 2045 0.8514536 FALSE
## 4 chr20.63863 chr20 63863 64424 13 562 0.8565943 FALSE
## 5 chr20.64609 chr20 64609 64728 6 120 0.6418476 FALSE
## 6 chr20.64778 chr20 64778 65365 6 588 0.1449167 FALSE
head(result$betas_per_segment)## Sample1 Sample2 Sample3 Sample4 Sample5 Sample6
## chr20.60008 0.7331288 0.7795786 0.8133333 0.84156977 0.7853659 0.81250000
## chr20.61141 0.8740920 0.7928571 0.8174157 0.86084906 0.8803612 0.89154013
## chr20.61818 0.8943894 0.8278388 0.7691083 0.89508197 0.8259587 0.82819383
## chr20.63863 0.8560748 0.8793103 0.8287154 0.87633262 0.7705628 0.81779661
## chr20.64609 0.5810056 0.7909605 0.5865385 0.63636364 0.6150943 0.71794872
## chr20.64778 0.2077922 0.1000000 0.2321429 0.02764977 0.0000000 0.07692308
## Sample7 Sample8 Sample9 Sample10 Sample11 Sample12
## chr20.60008 0.8653251 0.89727127 0.9042357 0.81441718 0.7964169 0.8934911
## chr20.61141 0.9152120 0.94017094 0.7426326 0.85650224 0.7752294 0.7946429
## chr20.61818 0.8787313 0.77701544 0.8374291 0.83053691 0.8900145 0.8762376
## chr20.63863 0.8160622 0.77941176 0.8909513 0.74315789 0.8958333 0.9621749
## chr20.64609 0.6912442 0.69565217 0.7898551 0.51933702 0.6346154 0.6311111
## chr20.64778 0.2844037 0.05769231 0.1984733 0.08024691 0.1188811 0.1530612
## Sample13 Sample14 Sample15 Sample16 Sample17 Sample18
## chr20.60008 0.8477930 0.85780526 0.8138848 0.8699187 0.8298755 0.89660743
## chr20.61141 0.9192140 0.91549296 0.8333333 0.8463357 0.8592965 0.78795812
## chr20.61818 0.8681481 0.81139756 0.8013468 0.8629283 0.8820059 0.93140244
## chr20.63863 0.8687783 0.83193277 0.8916479 0.9232614 0.8540434 0.80167015
## chr20.64609 0.5294118 0.66666667 0.6567164 0.4928910 0.6908213 0.65024631
## chr20.64778 0.1682692 0.05462185 0.2631579 0.1298077 0.2117117 0.05952381
## Sample19 Sample20
## chr20.60008 0.7971014 0.9366603
## chr20.61141 0.7953092 0.8556034
## chr20.61818 0.9240924 0.8153619
## chr20.63863 0.9120172 0.9628713
## chr20.64609 0.6808511 0.6701031
## chr20.64778 0.2166667 0.2110092